

In 2001, TAP Pharmaceutical Products was fined $875 million dollars1 by the United States Department of Justice. At the time, it was the largest fine in history against a pharmaceutical company. The company was charged with a number of infractions. But $290 million of the fine1 was levied for just one infraction: violation of the Prescription Drug Marketing Act (PDMA).
Enacted in 1987, the purpose of the PDMA is twofold2:
- To “…ensure that drug products purchased by consumers are safe and effective…”
- To “…avoid the unacceptable risk to American consumers from counterfeit, adulterated, misbranded, subpotent, or expired drugs.”
Since its enactment, the law has been vigorously enforced3, with violators fined billions of dollars, and some employees even sentenced to jail. Companies in the pharmaceutical industry will take whatever precautions necessary to avoid running afoul of the PDMA.
One indispensable (and government mandated) means of assuring PDMA compliance is through validated testing.
So What Is Validated Testing?
Simply put, validated testing is performed to assure that key systems comply with federally mandated requirements. For pharmaceutical companies, these requirements revolve primarily around the PDMA.
There are also some non-FDA regulations that mandate validated testing in other industries — notably privacy laws and the Sarbanes-Oxley Act (SOX), which mandates financial industry reform.
Today, I will focus on validation testing within the pharmaceutical industry.
Validated testing assesses systems rather than individual pieces of software. The goal, in the words of the FDA4, is “…to ensure accuracy, reliability, consistent intended performance, and the ability to discern invalid or altered records.”
Which Systems Should Be Validated?
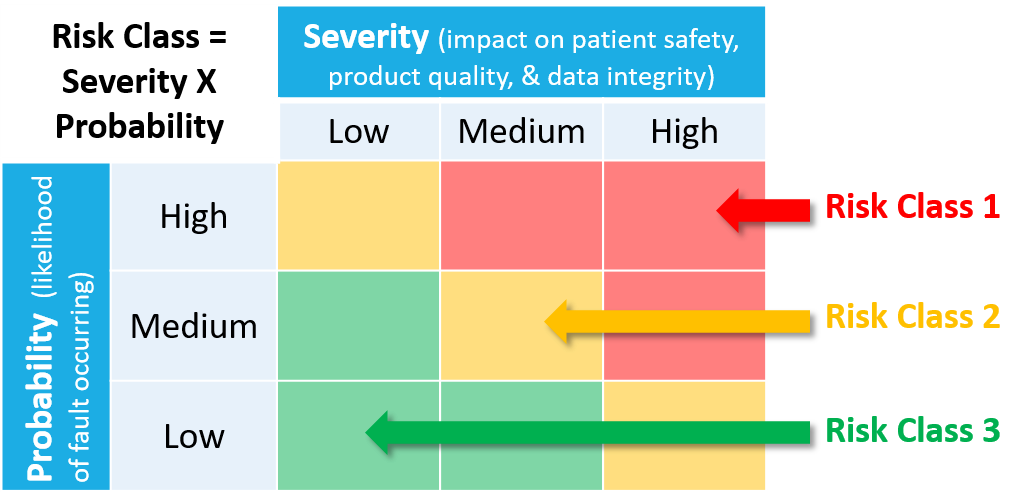
A risk-based approach is recommended to determine whether a system should be subject to validated testing. This approach examines the system and decides where it falls in a risk assessment, low (Risk Class 3), medium (Risk Class 2), or high (Risk Class 1).
The 3 risk classes can be defined as follows:
- Risk Class 1: This is a critical system (portrayed in red on the accompanying chart). The following systems should be placed in this risk class:
- Systems that have a direct impact on product quality, product efficacy, patient safety, or public health
- Systems that have a material impact on external financial reporting (SOW compliance)
- Systems that impact the privacy of patients, employees, health care providers, and consumers
- Risk Class 2: This is a non-critical system, but may still have a significant impact related to PDMA compliance (portrayed in yellow on the accompanying chart). The following systems should be placed in this risk class:
- Systems that have an indirect impact on product quality, product efficacy, patient safety, or public health
- Systems incorporating functionality that controls records required to meet regulatory requirements
- Risk Class 3: This is a non-critical system, with no impact to PDMA compliance (portrayed in green on the accompanying chart). The following systems should be placed in this risk class:
- Systems that have no impact on manufacturing, labeling, product testing, product quality, product efficacy, patient safety, or personnel safety
- Systems that have no impact on external financial reporting systems
- Systems that have no impact on regulatory requirements
The risk class assessment of a system is also impacted by the likelihood of a fault occurrence. The combination of severity + likelihood determines the final risk class assessment of a system.
What’s Different About Validation Testing?
Determining which systems require validation testing through the evaluation process detailed above is an important process. That’s because validation testing is a very involved, very controlled level of testing.
In other words, it’s expensive! Performing validation testing on a low-risk system doesn’t justify the expense and time required.
Essentially, validation testing encompasses 4 key areas:
- Validation Test Plan: The test plan for validation testing outlines the testing requirements and strategy. It should include the general process for performing the testing, documenting evidence of testing and the process for handling testing failures. The test plan should also include the types of testing, descriptions of environments where testing will be performed, who is responsible for testing, and equipment that will be used in testing.
- Comprehensive Requirements: The requirements documentation for validation testing must be extremely detailed. The requirements must detail exactly what we’re trying to accomplish. The requirements must be so specific as to leave no possibility of doubt or vagary about what is to be accomplished.
- Test Documents: The level of documentation is the largest differentiator between validation testing and ‘regular’ testing. The test documents must provide assurance that the system can consistently and repeatedly perform the various processes representing the intended use of the system.Required documentation will, at a minimum, include the following:
- Detail test scripts (often referred to as test protocols) that document each step of the process and provide expected results. The test protocols should be pre-approved by an independent software quality group.
- The test environment should be prepared:
- A test environment Installation Qualification (IQ) should be pre-approved, executed and post-approved
- The environment should be a controlled environment stable and not subject to change once testing has begun
- The system should be tested using comparable conditions and environmental characteristics to those of the production environment (i.e. mirror production environment as close as possible)
- Objective evidence that document the validity of results at each step of the test script (these are often screen shots). Objective evidence is data that shows or proves that something exists or is true, and others can objectively determine whether the conclusions are valid – “if it isn’t documented it didn’t happen”
- Post approval of the test scripts along with the objective evidence by an independent software quality group. Incident reports that document any failures or deviations from expected results
- Trace matrix report that links all test steps and test protocols back to original requirements documentation
- Prior to moving the code into Production, A system certificate should be drafted and a Production Installation Qualification (IQ) prepared and pre-approved.
- Detail test scripts (often referred to as test protocols) that document each step of the process and provide expected results. The test protocols should be pre-approved by an independent software quality group.
- Validation Summary Report: The Validation summary should provide a concise overview of the entire validation effort and the results obtained. The Validation Summary Report is used in conjunction with the System Certification to release the system for operational use.
The government is watching you…
The U.S. government is extremely serious about compliance of the PDMA. If a company operates systems that fall within the federal mandate for validation testing, it is essential that they follow the rules.
However, validation testing isn’t performed on the honor system. FDA representatives regularly audit companies that should be complying with the PDMA. Investigators visit companies, audit their records, and verify if they are compliant.
If the audit reflects that systems require validation testing, and the results of the testing are acceptable, then the company will pass the audit. However if documents suggest otherwise, the company can face an observation (slap on the wrist), fines, restrictions on the products they manufacture and distribute, and in extreme cases, imprisonment for accountable personnel.
Works Cited
1. United States Department of Justice (2001, Oct 3) "TAP PHARMACEUTICAL PRODUCTS INC. AND SEVEN OTHERS CHARGED WITH HEALTH CARE CRIMES; COMPANY AGREES TO PAY $875 MILLION TO SETTLE CHARGES" Retrieved from https://www.justice.gov/archive/opa/pr/2001/October/513civ.htm
2. FDA "Prescription Drug Marketing Act of 1987" Retrieved from
https://www.fda.gov/regulatory-information/selected-amendments-fdc-act/prescription-drug-marketing-act-1987
3. Pharmaceutical Online (2003, Sept 2) "CimQuest Launches New Hi-Q/PDMA™ Compliance Program as Strict Enforcement of Pharmaceutical Drug Marketing Act Hits Drug Firms with Billions of Dollars in Fines and Jail Terms" Retrieved from https://www.pharmaceuticalonline.com/doc/cimquest-launches-new-hi-qpdma-compliance-pro-0001
4. FDA https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=11.10